【昊文章】这么复杂的基因内重组引发疾病发生,你一定没见过
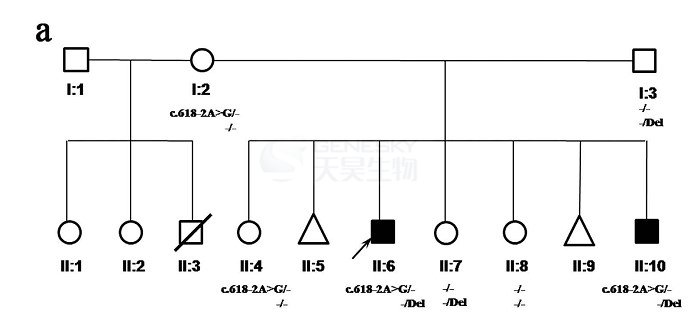
3月23日,《Scientific Reports》杂志报道了天昊客户有关Cockayne综合征家系致病位点的最新文章,题为A complex intragenic rearrangement ofERCC8 in Chinese siblings with Cockayne syndrome。该研究定位了一个中国CS家系的发病原因为ERCC8基因的复合杂合变异,包括了一个母源性的剪切位点变异,和一个父源性的引发外显子缺失的缺失/到位/缺失型复杂重组变异。
天昊生物参与完成了全外测序,基于全外数据的CNV分析,一代测序,天昊生物信息部李才华经理作为文章作者之一分享了研究成果。
本研究找到的基因内重组很复杂!!?不怕,先来看个知识点:
2007年底,CELL杂志报道了一种导致基因拷贝数变异的基因组重排新机制:细胞分裂的过程中分裂暂停,DNA复制转换了模板。这种新机制被称为“复制叉停滞和模板转换机制”(Fork Stalling and Template Switching,FoSTeS)。FoSTeS是由于错误的DNA复制而导致的不常见又特别复杂的一种重排机制,经常引起基因或基因内某区域的拷贝数变异。一段微同源(microhomology)序列是FoSTeS过程的一个重要特征。
该研究检测到的这种复杂的缺失/到位/缺失型复杂重组变异,即是来源于DNA断裂位置两侧微同源(microhomology)序列引发的2次“复制叉停滞和模板转换”(Fork Stalling and Template Switching,FoSTeS)现象。
文章解读
背景:
Cockayne综合征(CS;科凯恩氏综合征;MIM#133540,264400)是科凯恩在1936年首次发现的一种罕见常染色体隐性遗传疾病。该疾病涉及多个系统,其主要特征包括进行性发育迟滞,小头症,智力迟钝,视网膜色素变性,感觉神经性耳聋,光敏性和过早死亡。根据严重程度和表型异质性,临床上已经定义了多种CS亚型。例如,CS最经典的亚型—I型CS,在患者出生1-2年就会呈现发育异常,在10岁或20岁以内引发死亡。相比之下,II型CS更严重,出生时即出现症状, 而III型CS则症状较轻/症状晚发,即具有正常生长和认知发育。由于发病表型极其复杂多样,CS的精确诊断必须依靠基因检测。已知的CS致病基因包括:ERCC8突变(Cockayne综合征A或CSA,约占35%的CS病例)或ERCC6(Cockayne综合征B或CSB; 约占65%的CS病例)。
家系说明:
一中国CS家系,包括2名CS患者(黑色实心正方形框)。父母为非近亲婚配。2例患者已完成ERCC8/ERCC6测序,但并没有找到纯合突变或复合杂合致病突变,但提示可能存在一个影响ERCC8剪切的突变。父母也接受了核型检测和cCGH检测,未发现可疑致病基因组不平衡或单亲二倍体现象。
技术平台:
全外显子组测序;比较基因组芯片;qPCR,一代测序
结果:
1. 对2例患者进行全外显子测序,基于以下思路筛选出唯一的可能致病位点:ERCC8c.618-2A > G,该变异来源于患者母亲。
外显子测序数据---正常样本公共数据库过滤----2例患者共有罕见变异826个--- Ingenuity Pathways Analysis (IPA)软件进行表型-基因型分析----依据美国医学遗传学和基因组学学院(ACMG)变异指南筛选致病变异---在家系中与表型共分离--- 一个可变剪切区变异(ERCC8 c.618-2A > G)
2. 基于患者ERCC8 cDNA测序,确认该变异的确导致了ERCC8基因8号外显子的前9 bp (TGCTGACAG)序列缺失,从而编码出缺失三个氨基酸的ERCC8蛋白。
3. 由于一个正常的同胞(患者妹妹)也携带该突变,因此,研究者猜测可能存在另一个缺失、插入型的致病突变。基于每个外显子Q-PCR, 1 M aCGH检测,长PCR测序,最终确定了2名患者都携带一个引发4号外显子丢失的3.8kb的缺失,该缺失变异来源于患者父亲。
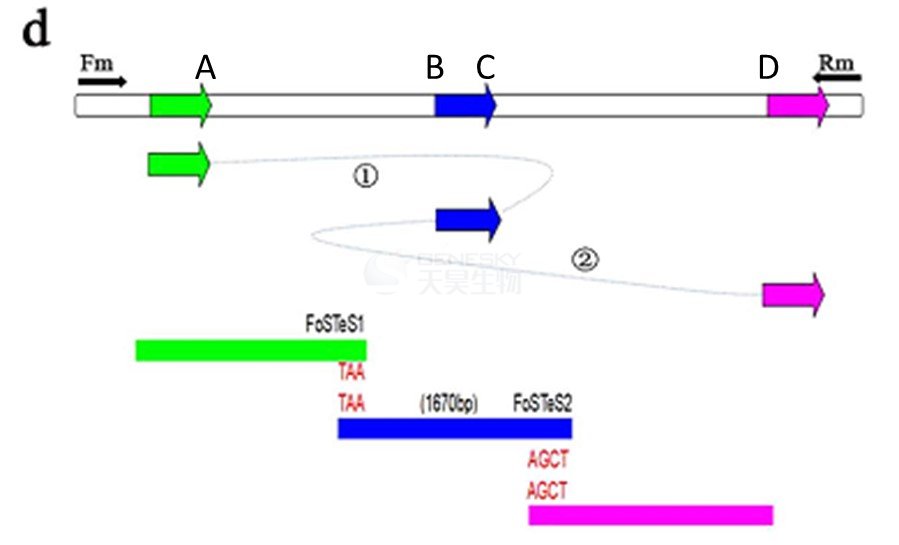
4. 基于PCR产物胶回收和一代测序,确认该缺失实际上包括2个缺失片段和1个1.67kb片段倒位(位于2个缺失片段中间的)。
AB, CD之间的序列为缺失片段,但由于A、C处的序列都为TAA, B、D处的序列都为AGCT,即所谓的DNA断裂位置两侧微同源(microhomology)序列,所以片段1和片段2缺失后,中间的1.67kb序列发生了到位,A与C连接,B与D连接。这种复杂的重组机制,有可能是由DNA断裂位置两侧微同源(microhomology)序列引发了2次“复制叉停滞和模板转换”(Fork Stalling and Template Switching,FoSTeS)。
结论: 该研究定位了一个中国CS家系的发病原因为ERCC8基因的复合杂合变异,其中一个变异为来源于母亲的剪切位点变异,另一个为来源于父亲的引发外显子缺失的缺失/到位/缺失型复杂DNA重组。
这么精彩的故事,如果你也想讲一个,那么天昊生物可以提供的是:
-
众多项目和文献支持的全外显子测序技术平台
-
基于最新流程的外显子测序数据后的基因组SNV和CNV同步分析
-
专业的团队:天昊生物信息分析和实验团队,大多数为医学遗传学专业背景,在人类医学研究方面拥有丰富的生物信息和遗传统计分析经验
-
最严格质控:天昊具备整套的实验流程质控,利用天昊特有的SNPscan 96 SNP分型质控体系确保二代测序样品及数据质量的准确性。针对大样本二代测序实验样本易混的风险,独创分子内
更多天昊部分客户基因组测序文章:
|
Xu, F., et al., Whole-exome and targeted sequencing identify ROBO1 and ROBO2 mutations as progression-related drivers in myelodysplastic syndromes. Nat Commun, 2015. 6: p. 8806. |
|
Zhang, Z., et al., Exome sequencing identifies SLCO2A1 mutations as a cause of primary hypertrophic osteoarthropathy. Am J Hum Genet, 2012. 90(1): p. 125-32. |
|
Zhang, Z., et al., Genomic variations of the mevalonate pathway in porokeratosis. Elife, 2015. 4: p. e06322. |
|
Xiao, H., et al., Mutations in epigenetic regulators are involved in acute lymphoblastic leukemia relapse following allogeneic hematopoietic stem cell transplantation. Oncotarget, 2016. 7(3): p. 2696-708. |
|
Zheng, Z., et al., Molecular defects identified by whole exome sequencing in a child with Fanconi anemia. Gene, 2013. 530(2): p. 295-300. |
|
Yang, X., et al., Mutational analysis of CHCHD2 in Chinese patients with multiple system atrophy and amyotrophic lateral sclerosis. Journal of the Neurological Sciences, 2016. 368: p. 389. |
|
Qiu, Y.L., et al., Defects in MYO5B are associated with a spectrum of previously undiagnosed low γ‐glutamyltransferase cholestasis. Hepatology, 2016. |
|
Luan, X., et al., Infantile spinal muscular atrophy with respiratory distress type I presenting without respiratory involvement: Novel mutations and review of the literature. Brain & Development, 2016. 38(7): p. 685. |
|
Mei, R., et al., Exome sequencing identifies a novel mutation in GJA8 associated with inherited cataract in a Chinese family. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie, 2016. 255(1): p. 141-151. |
|
Jiang, T., et al., A rare coding variant in TREM2 increases risk for Alzheimer's disease in Han Chinese. Neurobiology of Aging, 2016. 42: p. 217.e1. |
|
Li, J.J., et al., Familial Hypercholesterolemia Phenotype in Chinese Patients Undergoing Coronary Angiography. 2016: p. ATVBAHA.116.308456. |
|
Miao, C., et al., Mutations in the Motile Cilia GeneDNAAF1Are Associated with Neural Tube Defects in Humans. G3 Genesgenetics, 2016. 6(10): p. 3307-3316. |
|
Li, T., et al., Identification of candidate genes for congenital heart defects on proximal chromosome 8p. Scientific Reports, 2016. 6: p. 36133. |
|
Zhang, Z.L., et al., The identification of novel mutations in COL1A1 , COL1A2 , and LEPRE1 genes in Chinese patients with osteogenesis imperfecta. Journal of Bone and Mineral Metabolism, 2012. 30(1): p. 69-77. |
 咨询热线:400-065-6886
咨询热线:400-065-6886