◆先天性肝纤维化(CHF)为一种罕见的先天发育异常,其主要后遗症是门静脉高压症(PH),临床上常表现为脾肿大、脾功能亢进和静脉曲张出血。CHF反映了纤毛原发性的功能障碍,并常伴有其他器官畸形或其他系统的故障。大多数存在DPM/CHF的纤毛疾病只有在并发感染或胆道梗阻时才会表现出胆汁淤积症。
◆肝内胆汁淤积症可以分为2类:一类是血清中γ-谷氨酰转移酶(GGT)的活性与血清共轭胆红素都上升;另一类是在正常范围内,但存在高胆红素血症。
◆对于许多临床表型为肝内胆汁淤积症和高GGT活性的儿童患者,其肝胆病的遗传病因仍未确定。本研究的目标是为了发现患有特发性高GGT肝内胆汁淤积症儿童的疾病相关突变基因。
样本收集:患者:2004-2017年,纳入儿童患者25例,均表现原因不明的肝内胆汁淤积症和高GGT。对照样本:其它肝病患者对照(45例,GGT正常的胆汁淤积患者或者无胆汁淤积的其它肝病患者);无肝脏疾病的对照样本(299例)。
实验方法:WES测序;mRNA和蛋白序列分析;免疫组化检测;表达载体构建;基因敲降;从患者诱导的多能细胞中提取成纤维细胞样细胞;免疫荧光显微镜分析;免疫蛋白印迹
● ZFYVE19中的双等位基因突变与高GGT肝内胆汁淤积症有关
基于25例患者的WES或目的区域测序数据,有9例患者都存在ZFYVE19纯合或复合杂合突变(图1AB;P1患者因DNA不足,采取ZFYVE19目的区域测序,其它患者为WES测序样本),家系样本验证确定突变的遗传模式是常染色体隐性,且患者均没有ABCB4突变。其它两个对照人群均没有ZFYVE19的双等位基因突变。
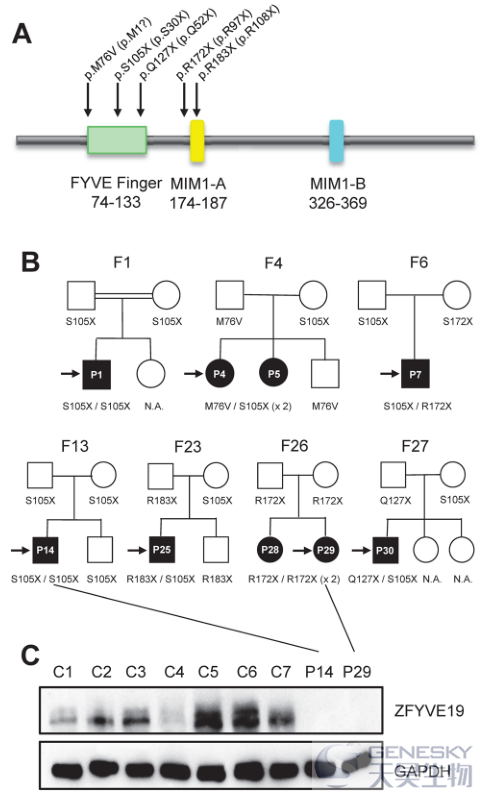
图1:9例患者检测到的5个ZFYVE19突变和患者肝组织的蛋白表达水平
● ZFYVE19基因的备用翻译起始位点
在ZFYVE19基因富含鸟嘌呤的区域发现了G-四联体(图2A)。数据库注释的翻译起始位点AUG很可能被G-四联体的存在所抑制(G-四联体是稳定的翻译抑制性二级结构,大多位于5′非翻译区域)。
c.112_113insGGGGC(rs142730574)为一个无功能多态,在ExAC中记录的频率很高,杂合子为35.55%,同源子为12.6%。当GGGGC存在时,下游具有Kozak序列的AUG(dAUG)可能是一个替代性的翻译启动点,可以从76位蛋氨酸(M76)开始翻译出一个396个氨基酸的的异构体。M76在不同物种中具有高保守性(图2B)。基于体外过表达实验,证明当c.112_113insGGGGC存在时,破坏了471个氨基酸长度的ZFYVE19蛋白生成,只能生成396个氨基酸的异构体。针对这个异构体,重新注释之前在患者中测到的突变,5个突变均完全破坏这个异构体的功能。
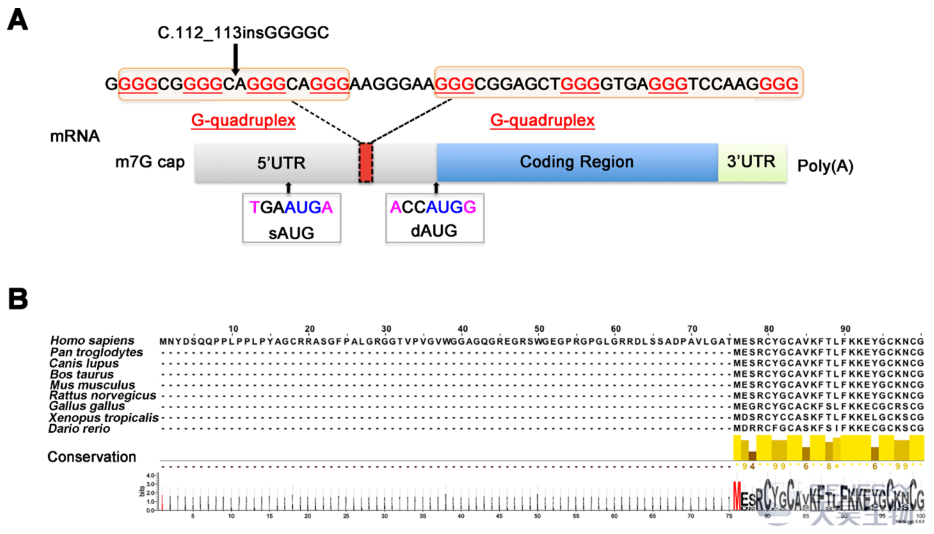
图2:ZFYVE19的mRNA序列和M76的保守性
● ZFYVE19敲降产生的表型导致纤毛和中心粒异常
患者肝组织的组织病理学和免疫组织化学研究结果均提示存在纤毛病理,作者评估了hRPE1和成纤维细胞中ZFYVE19敲降后的纤毛相关表型。在ZFYVE19基因敲降的hRPE1细胞中,一个突出的表型异常是基底体/中心粒的数量增加。同时也观察到中心粒对的分离/不正常排列(图5A)。纤毛的组装没有受到影响(图5C),额外的纤毛在额外的基体/中心粒处形成(图5A)。源自患者iPSCs(图5D-F)的ZFYVE19缺失成纤维细胞样细胞中,也观察到类似表型异常。因此,研究者的数据表明ZFYVE19参与了纤毛相关过程。
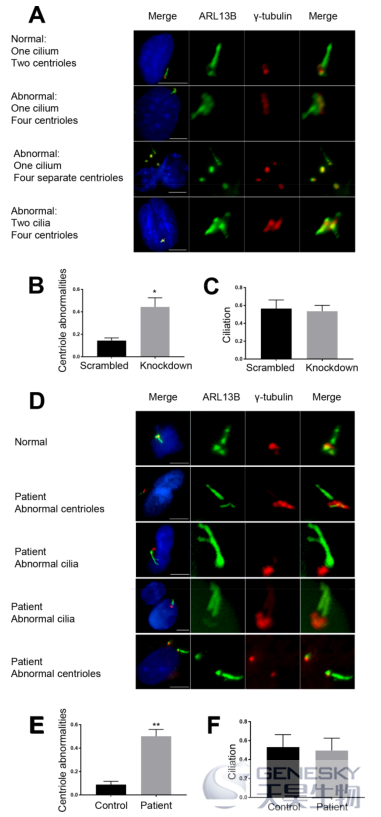
图5:ZFYVE19敲降产生的表型导致纤毛和中心粒异常
●鉴定出ZFYVE19基因存在下游翻译起始密码子,促使研究者证明ZFYVE19存在起始于M76的功能异构体。在ZFYVE19患者中发现的五个突变都位于介导ZFYVE19和囊泡蛋白分选相关蛋白4(VPS4)相互作用的FYVE型锌指域或MIM1-A基团。五个突变被预测存在致病性,很可能会导致翻译启动被破坏或者RNA的无意降解,导致ZFYVE19完全丧失功能。事实上,在携带纯合无义突变的患者(P14和P29)中,研究者的确没有检测到ZFYVE19在肝脏中的表达(图1C)。
●纤毛功能障碍引起的发育和退行性疾病,被称为纤毛疾病,影响到各个器官和系统,并表现出不同的临床特征。在肝脏中,肝细胞不表达纤毛,但胆管细胞表达。它们的初级纤毛延伸到胆管腔内。胆汁的变化流动和组成被认为是通过纤毛感知并转化为功能的变化。
编码纤毛相关蛋白的基因突变可能会导致门静脉的发育不全,桡骨,门静脉纤维化增加和增生。这一系列异常被称为管板畸形(DPM)或CHF。本研究的ZFYVE19突变患者在组织病理上的表现与DPM或CHF一致,同时伴有硬化性胆管病。我们认为他们的疾病是一种新的纤毛病,并将其归因于ZFYVE19的突变。
●然而,本文探讨的遗传性肝病有一个临床表型并不常见于纤毛病。表现为DPM或CHF的纤毛病一般与GGT值的升高和胆汁淤积症(除非胆道梗阻或感染)无关。此外,本文的患者有硬化性胆管病与胆汁淤积的组织病理特征。这些变化表明胆道损伤是ZFYVE19肝病的重要表现。
●另外值得关注的是,本文的患者临床上缺乏肝外表现。纤毛疾病,理论上是一种多器官和多系统的疾病。研究者预计,随着对其他ZFYVE19突变患者的研究,疾病表型将扩展到肝胆道以外的器官和系统。此外,研究者还将对ZFYVE19疾病患者进行长期跟踪。看看随着他们的成长,ZFYVE19缺失是否会产生新的影响。鉴于体外实验发现ZFYVE19敲降后发生异常的染色体分离和DNA损伤,研究者将考虑恶性肿瘤风险增加的可能性。
探索真理的道路从来不是一帆风顺,创新性越强,研究难度越大,耗时越长。从初次通过二代测序方法从病人中筛选出ZFYVE19,到研究成果被认可并发表,历时漫长的6年时间,经历了多位研究团队成员的接力奉献。由于既往对于这一基因功能的研究十分有限,初期在国际学术会议进行报道时受到了业内学者的较多质疑。为了寻找致病证据,课题组研究人员积极收集更多病例、随访病人,提供具有说服力的家系证据;与华山医院、仁济医院合作,收集病人肝移植术中的组织样本进行研究,证明在病人中该蛋白表达缺失;与奥地利格拉茨大学医学院病理系合作,分析研究病人肝组织的病理特征,从病理特征推测此病应该是纤毛病,终于找到进一步深入研究的线索;与中科院深圳先进技术研究院合作,通过细胞系模型和病人来源的诱导干细胞实验证实ZFYVE19变异影响纤毛形成,首次明确该基因与纤毛形成的直接相关性;与复旦大学生物医学研究院合作,发现ZFYVE19基因的数据库注释存在缺陷,从而导致这一基因在测序分析中容易被忽略,作者通过该基因转录和蛋白翻译方面进行深入研究,对该基因的翻译起始位点进行了新的注释,故而至今才被学术界揭示,克服了注释错误对研究造成了多重壁垒。在多方协助、共同努力下,课题组克服重重困难,历时多年的研究成果终得以获得业内专家认可,也代表复旦大学附属儿科医院肝病科的专业研究水平登上了一个新的高峰。
更多天昊WES平台高分文章链接:
【昊文章】天昊WES 平台文章新高度,助力中国学者见刊Neuron;
【昊文章】《Brain》新文:基于闽南地区遗传性痉挛性截瘫(HSP)家系测序与功能研究确定HSP的新致病基因;
兼程并进!天昊助力男性不育多中心研究者确定引发男性不育的两个新致病基因
咨询沟通请联系
18964693703(微信同号)

创新基因科技,成就科学梦想
 咨询热线:400-065-6886
咨询热线:400-065-6886



前往“发现”-“看一看”浏览“朋友在看”